Depressants
Step 1: What the Drug Does
Depressants are not one mechanism. They all end at "more inhibition," but they get there through different routes. They inhibit through increasing GABA (inhibitory) neuron activity or decreasing glutamate (excitatory) neuron activity.
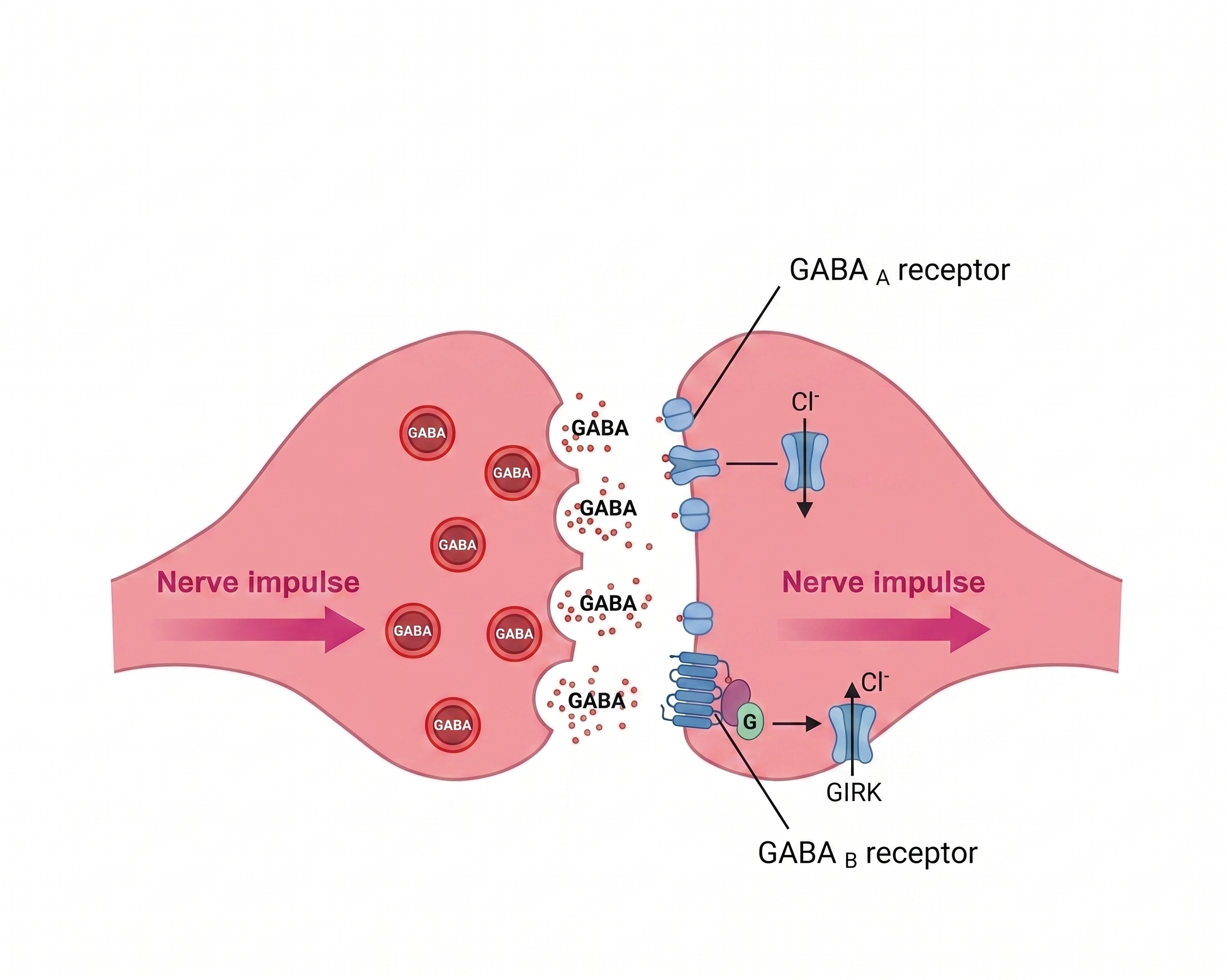
GABA-A Positive Allosteric Modulators (PAMs)
Drugs: benzodiazepines, Z-drugs.
Don't activate the receptor on their own. Bind a different (allosteric) site on the GABA-A receptor and amplify whatever GABA is already doing. Increase the frequency of channel opening when GABA is present (Goldschen-Ohm, 2022).
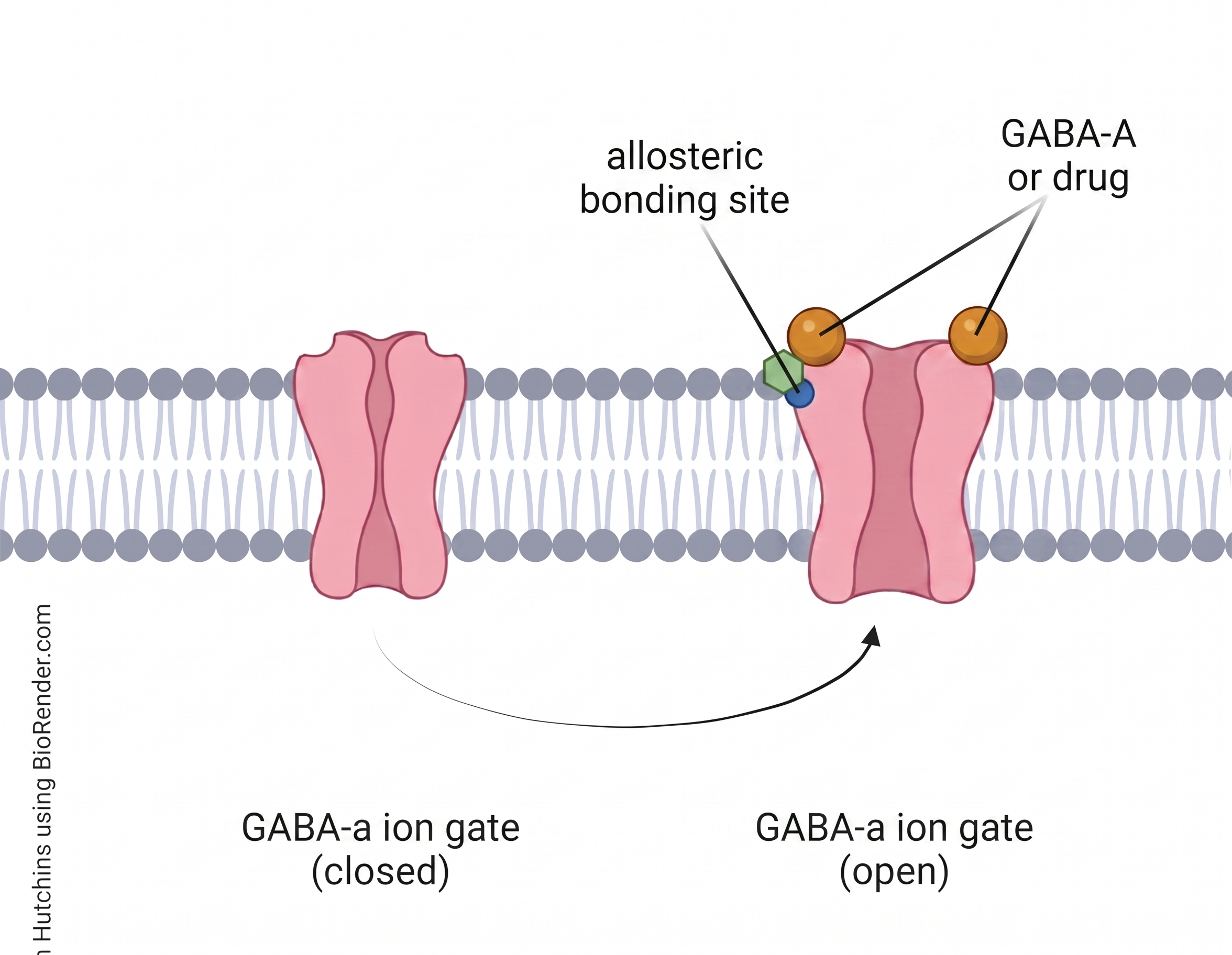
Result: more Cl⁻ into cell → hyperpolarized → less likely to fire.
Crucial detail: benzodiazepines have a ceiling. Cannot open the channel without GABA. Only enhance natural signaling. This is why pure benzo overdose is rarely fatal on its own.
Video: Benzodiazepines - click to expand ↗ YouTube
Barbiturates - Direct GABA-A Channel Openers
Drugs: phenobarbital, secobarbital, pentobarbital.
True direct channel openers that can activate GABA-A even without GABA present. No ceiling. This is the mechanistic reason barbiturate overdose is acutely lethal at lower doses than alcohol, and why they were largely replaced by benzodiazepines in clinical use (Macdonald & Kelly, 1995).
GABA-B Agonists
Drugs: GHB, phenibut, baclofen.
GABA-B is a separate receptor from GABA-A. It's a G-protein-coupled receptor that opens potassium channels and closes calcium channels. These drugs directly agonize GABA-B → strong sedation, euphoria, muscle relaxation (Lapin, 2001).
Phenibut also has α2δ calcium channel activity at higher doses (see Type 4). GHB has additional effects at its own dedicated GHB receptor.
α2δ Calcium Channel Modulators
Drugs: gabapentin, pregabalin (Lyrica), phenibut (partially).
These do not bind GABA receptors at all, despite their names. They bind the α2δ subunit of voltage-gated calcium channels on presynaptic terminals. This reduces calcium influx when the neuron tries to fire → reduces release of excitatory neurotransmitters (glutamate, substance P, norepinephrine).
Net effect resembles GABAergic inhibition. They suppress excitation rather than amplify inhibition (Imani & Rahimzadeh, 2012).
This is why gabapentin and pregabalin are dosed in hundreds of milligrams while benzos are dosed in single milligrams. Different molecular targets, different potencies. Dependency risk is lower and they are less dangerous but also less euphoric.
Step 2: What GABA Normally Does
GABA is the brain's main inhibitory neurotransmitter - roughly 20–40% of all brain synapses are GABAergic. Its function is the neural brake: when GABA binds its receptors, it makes the receiving neuron harder to fire.
Two receptor types carry this signal:
- GABA-A - fast. A chloride channel. GABA binding opens the channel → Cl⁻ rushes in → hyperpolarization. This is the target for most depressants.
- GABA-B - slow. G-protein coupled. Opens K⁺ channels and closes Ca²⁺ channels. Same inhibitory end result through slower, longer-lasting machinery.
GABA is ubiquitous: cortex, amygdala, cerebellum, brainstem, reward circuits. That breadth is why depressants touch nearly every brain function at once.
Step 3: Where GABA Lives
Each region's normal job is what gets altered by depressant use. See Step 4 for how.

 img.png)



Step 4: How Side Effects Fall Out (Types 1–3)
Each side effect = GABA amplified (or glutamate suppressed) in a specific region altering that region's normal job.
Sedation, Slowed Thinking, Slurred Speech
Cortex GABA amplified → cortical neurons inhibited → conscious processing slowed. The same mechanism that reduces anxiety also dims cognition and motor speech at higher doses.
↓
Anxiolysis (Anxiety Reduction)
Amygdala GABA amplified → fear-processing neurons silenced → anxiety drops. This is the therapeutic effect and the primary reason for abuse.
↓
Disinhibition / "Drunk" Behavior
The prefrontal cortex (PFC) is inhibited first, being most sensitive to GABA enhancement. The brainstem is inhibited last. The brake on impulsive behavior comes off while the user is still conscious and mobile. Judgment, social filters, and risk assessment fail before motor control does.
 ↓
↓
Anterograde Amnesia / Blackouts
Hippocampal LTP requires functional excitatory transmission. Suppress that transmission → new memories don't form. The user appears awake and functional but is not encoding the experience. This is not a gap in recall. The memory was never made.
↓
Ataxia (Loss of Motor Coordination)
Cerebellum GABA amplified → motor coordination circuits inhibited. Why drunk people stumble. The cerebellum integrates movement sequences. When it's suppressed, actions become uncoordinated and imprecise.
↓
Euphoria
Same disinhibition logic as opioids - VTA GABAergic interneurons normally brake dopamine release. Amplify GABA on those GABA neurons → ↓ the brake → ↑ dopamine in NAc. GABA on GABA on dopamine.
↑
Respiratory Depression - what kills
Medullary respiratory centers' GABA-A and GABA-B receptors over-amplified → breathing-rhythm neurons hyperpolarized → breathing slows. Gabapentinoids contribute via reduced excitatory drive to those same neurons. Mixing depressants with opioids is especially dangerous: two independent mechanisms suppressing the same circuit.
↓
Mixing depressants with different mechanisms can produce effects greater than the sum of the parts. Each drug suppresses a different node in the same circuit.
Specific Drug Notes
GHB ›
Near-vertical dose-response curve. The dose that produces euphoria and the dose that produces unconsciousness or death are very close. Sold in liquid form with inconsistent concentration. Misjudgment by milliliters can kill.
Phenibut ›
Sold as a "supplement" and legally available in many places. Produces tolerance fast. Withdrawal is severe and comparable to benzodiazepine withdrawal, including seizure risk. Many users underestimate it because it is not a controlled substance.
Gabapentin & Pregabalin ›
Not classically scheduled in many jurisdictions, but produce real dependence and dangerous withdrawal. Pregabalin is now a major recreational drug in parts of Europe and the UK. They are not "weak benzos." Different target, different overdose risk profile.
Alcohol ›
Mechanism: PAM + NMDA Antagonist. Alcohol is a positive allosteric modulator (PAM), not a direct agonist - but it binds different GABA-A subunit combinations than benzodiazepines do. Benzos target γ-subunit-containing synaptic receptors; alcohol preferentially potentiates δ-subunit-containing extrasynaptic receptors (cerebellar granule cells, thalamus). Same general outcome, different receptor populations.
Alcohol also significantly antagonizes NMDA receptors - it is a dual-mechanism depressant. At very high concentrations it can have some direct channel-opening activity, but at drinking doses the mechanism is PAM + NMDA antagonism, not direct channel opening. It also hits glycine receptors, voltage-gated calcium channels, and serotonin systems simultaneously - this breadth is why alcohol withdrawal is more medically dangerous than withdrawal from any single GABAergic drug (Zhu et al., 2007).
Why it kills more readily than benzos: The extrasynaptic GABA-A potentiation has a different ceiling profile than the benzo/synaptic mechanism, and NMDA antagonism adds a second independent route of CNS depression. Neither route has the benzo's protective ceiling.
The Balancing Loop
Continuous depression of activity → the brain compensates by:
- Downregulating GABA-A receptors (especially the α1 subunit) - fewer receptors means less sensitivity to the drug (Foitzick et al., 2020).
- Upregulating glutamate (NMDA) receptors to push back against the suppressed excitation.
- For α2δ drugs: changes in calcium channel expression and synaptic plasticity.

The withdrawal mechanism is the same across the class even when the acute mechanisms differ. Whether the brain compensated for an alcohol-driven Cl⁻ influx, a benzo-amplified GABA signal, a GHB hit on GABA-B, or a gabapentin-blocked calcium channel. When the drug leaves, the compensations are still in place. The brain is now tilted toward excitation, with too few brakes (Vinkers & Olivier, 2012).
User Manual
Abrupt withdrawal from alcohol, benzodiazepines, GHB, phenibut, and gabapentinoids can kill. This is rare among drug classes. Unopposed glutamate hyperexcitability → status epilepticus, cardiovascular collapse, hyperthermia.
Cross-tolerance is real within the class. A person tolerant to alcohol is partially tolerant to benzos. A person tolerant to phenibut is partially tolerant to GABA-B medications. This is the basis for medical detox - replacing one depressant (e.g., alcohol) with a long-acting one (e.g., diazepam or chlordiazepoxide) that can be tapered safely.
Cross-tolerance does NOT extend to opioids. A person with high depressant tolerance can still die from a standard opioid dose - they operate on different receptors.
User Manual
- Never stop benzodiazepines, heavy alcohol use, GHB, phenibut, or pregabalin/gabapentin cold turkey after sustained use. Taper under medical supervision.
- Never combine depressants with opioids. They suppress the same breathing circuit through independent mechanisms. There is no shared ceiling. The combined effect is multiplicative, not additive.
- Do not mix depressants from different mechanism classes (e.g., benzodiazepines + alcohol). Benzos amplify GABA-A signaling at the allosteric site; alcohol potentiates a different subset of GABA-A receptors (δ-subunit extrasynaptic) and simultaneously antagonizes NMDA receptors. Two overlapping but distinct routes of CNS depression combine. Neither has the other's safety ceiling. The result is unpredictable, potentially multiplicative respiratory depression.
- For drug-specific safety notes, see the expandable sections above.
- Never use depressants alone if using at doses where unconsciousness is a risk. Respiratory depression from CNS depressants can render you unable to call for help. Have someone present or nearby with naloxone accessible if opioids are also involved.
Sources
- Brunton, L. L., Hilal-Dandan, R., & Knollmann, B. C. (Eds.). (2017). Goodman & Gilman's: The Pharmacological Basis of Therapeutics (13th ed.). McGraw-Hill Education. https://accessmedicine.mhmedical.com/content.aspx?bookid=2189§ionid=165936845
- Foitzick, M. F., Medina, N. B., Iglesias García, L. C., & Gravielle, M. C. (2020). Benzodiazepine exposure induces transcriptional down-regulation of GABAA receptor α1 subunit gene via L-type voltage-gated calcium channel activation in rat cerebrocortical neurons. Neuroscience Letters, 721, 134801. https://doi.org/10.1016/j.neulet.2020.134801
- Goldschen-Ohm, M. P. (2022). Benzodiazepine modulation of GABA-A receptors: a mechanistic perspective. Biomolecules, 12(8), 1467. https://doi.org/10.3390/biom12081467
- Imani, F., & Rahimzadeh, P. (2012). Gabapentinoids: gabapentin and pregabalin for postoperative pain management. Anesthesiology and Pain Medicine, 2(2), 52–53. https://doi.org/10.5812/aapm.7743
- Lapin, I. (2001). Phenibut (β-phenyl-GABA): a tranquilizer and nootropic drug. CNS Drug Reviews, 7(4), 471–481. https://doi.org/10.1111/j.1527-3458.2001.tb00211.x
- Macdonald, R. L., & Kelly, K. M. (1995). Antiepileptic drug mechanisms of action. Epilepsia, 36(Suppl 2), S2–S12. https://doi.org/10.1111/j.1528-1157.1995.tb05996.x
- Neuroscientifically Challenged. (n.d.). 2-Minute neuroscience: Benzodiazepines [Video]. YouTube. https://www.youtube.com/watch?v=D5Vsm_Daexg
- Sarkar, D. K., & Boyadjieva, N. I. (2007). Ethanol alters production and secretion of estrogen-regulated growth factors that control prolactin-secreting tumors in the pituitary. Alcoholism: Clinical and Experimental Research, 31(12), 2101–2105. https://doi.org/10.1111/j.1530-0277.2007.00539.x
- Vinkers, C. H., & Olivier, B. (2012). Mechanisms underlying tolerance after long-term benzodiazepine use: a future for subtype-selective GABA-A receptor modulators? Advances in Pharmacological and Pharmaceutical Sciences, 2012, 416864. https://doi.org/10.1155/2012/416864
- Zhu, W., Bie, B., & Pan, Z. Z. (2007). Involvement of non-NMDA glutamate receptors in central amygdala in synaptic actions of ethanol and ethanol-induced reward behavior. The Journal of Neuroscience, 27(2), 289–298. https://doi.org/10.1523/JNEUROSCI.3912-06.2007